 |
| Visit our sponsor at www.chemglass.com |
|
| Visit our sponsor at www.chemglass.com |
 |   |
| Fischer Carbene Complexes |
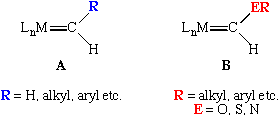
These are sometimes called Fischer carbenes in honor of E.O. Fischer, who reported the first example in 1964 and later won a Nobel Prize for his pioneering work on ferrocene with Wilkinson.
Fischer carbenes are typically found on electron-rich, low oxidation state metal complexes (mid to late transition metals) containing pi-acceptor ligands. The presence of the heteroatom on the alpha carbon allows us to draw a resonance structure that is not possible for an unsubstituted (Schrock-type) alkylidene:
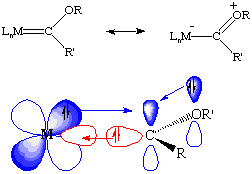
If we look at this from a molecular/atomic orbital perspective, one lone pair is donated from the singlet carbene to an empty d-orbital on the metal (red), and a lone pair is back-donated from a filled metal orbital into a vacant pz orbital on carbon (blue). There is competition for this vacant orbital by the lone pair(s) on the heteroatom, consistent with our second resonance structure. Overall, the bonding closely resembles that of carbon monoxide. Therefore, carbene ligands are usually thought of as neutral species, unlike dianionic Schrock alkylidenes (which usually lack electrons for back-donation). But remember that electron counting is just a formalism!
The stabilization of a partial positive charge on the carbene ligand has predictable consequences that show up in the reactivity and spectroscopic features (see below).
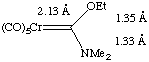
The stronger the pi-donor on the carbene carbon, the lower the M=C bond order and lower the barrier to rotation around the M-C bond.
Proton and carbon NMR data are similar to those observed for alkylidene complexes, however, Fischer carbenes do not typically display any unusual JCH values that might indicate an agostic interaction between the carbene proton and the metal.
Fischer carbenes often contain carbonyl ligands which can provide very useful NMR and IR data.
There are many synthetic methods for the synthesis of carbene complexes. The four most common ones are shown here.
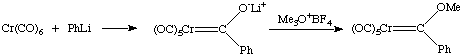
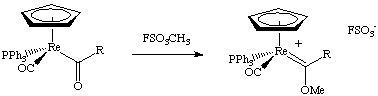
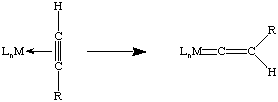
Acetylides can be easily synthesized from Li or SiMe3 acetylides. These species are quite reactive towards electrophiles at the beta carbon, i.e. they are easily protonated or alkylated:
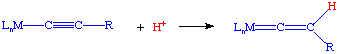
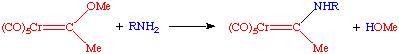
These reactions go in good yield and are stereospecific. A second example is by Helquist et al. J. Am. Chem. Soc. 1982, 104, 1869:
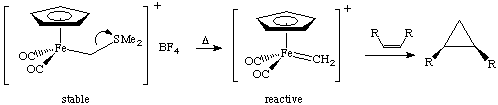
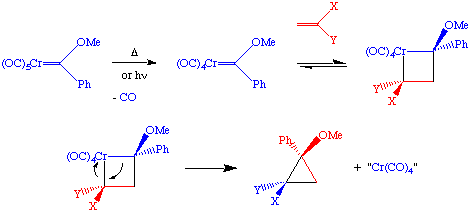
Substituting a phosphine for one CO in the above reaction leads to good enantiomeric excess. One should note that there is well-developed cyclopropanation chemistry using diazoalkanes (explosive!) as the carbene source; organometallic reagents are much safer.
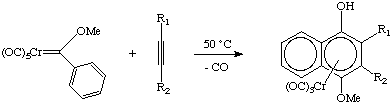
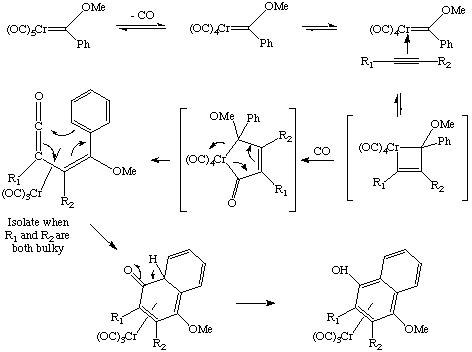
The metal can be easily cleaved from the naphthol. Dötz-type chemistry is well-developed and has been used to prepare Vitamin E as well as some antibiotics. What is the mechanism of this "termolecular" reaction? Experiments show that the rate is inhibited by excess CO or phosphine, so the first step must be dissociation of CO. Coordination of the alkyne, formation of a metallacyclobutene, insertion of CO into the ring, rearrangement to a pi-complex, ring closure and a proton shift account for our observed product (whew!):

[Index] [Keyword Search] [Books & Software] [ILPI Home Page]
Please visit our sponsor to thank them for supporting this site!
This page was last updated Tuesday, March 31, 2015
This document and associated figures are copyright 1996-2025 by Rob Toreki or the contributing author (if any) noted above. Send comments, kudos and suggestions to us by email. All rights reserved.